Das Universum der Moleküle
Für die Suche nach neuen Medikamenten haben Chemiker praktisch alle theoretisch möglichen Moleküle bis zu einer gewissen Grösse berechnet. Um sich in der astronomischen Informationsfülle zurechtzufinden, haben sie dafür auch noch eigene Navigationssysteme entwickelt.
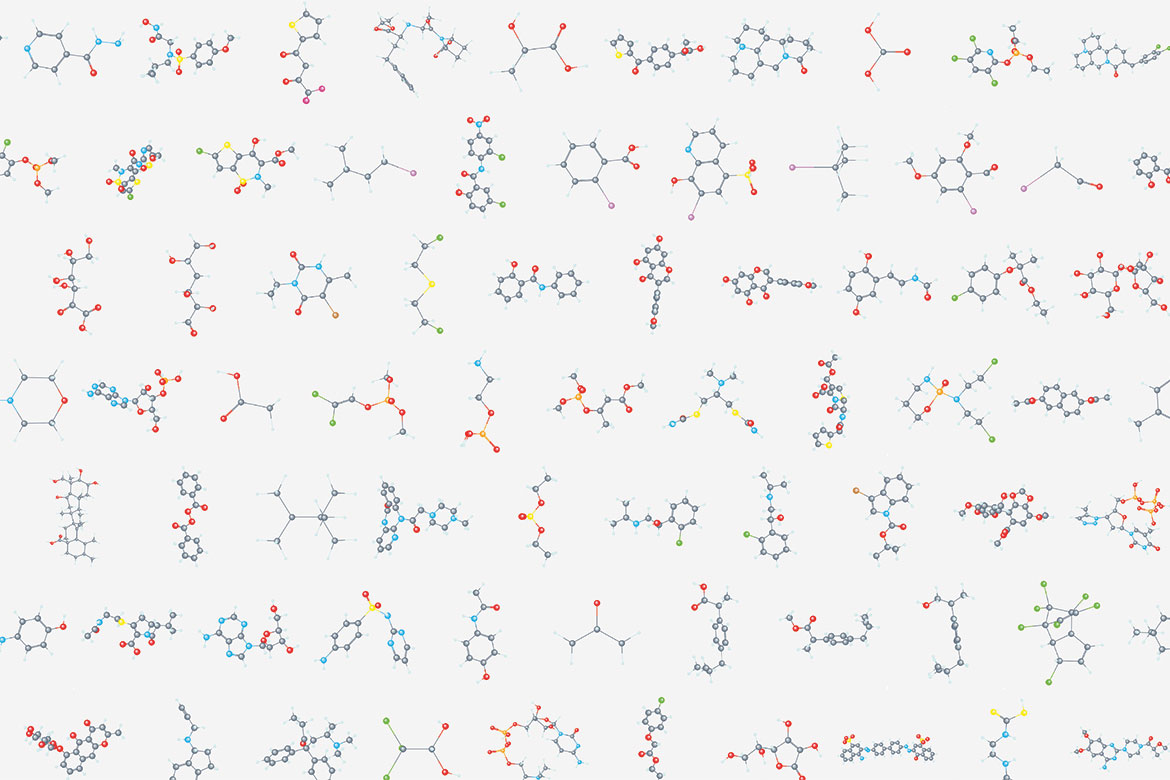
Die Zahl der theoretisch möglichen Moleküle ist grösser als die Zahl aller Atome im Kosmos. Auf dem Bild ist ein Bruchteil davon sichtbar. | Bild: 2. stock süd
Wie viele Moleküle können überhaupt existieren? Anfangs habe ihn die rein akademische Frage interessiert, erzählt Jean-Louis Reymond von der Universität Bern. Der Chemiker wollte wissen, wie viele Moleküle bekannt sind und wie viele neue, unbekannte es noch geben könnte. Im Jahr 2001 begann er mit seinem Team Moleküle zu zählen und in gigantischen Datenbanken systematisch zu sammeln. Er wollte neue Wirkstoffe finden, mit denen sich schwere Krankheiten heilen lassen.
So entwickelte Reymond numerische Methoden, um praktisch alle theoretisch möglichen Moleküle bis zu einer gewissen Grösse zu berechnen und deren Eigenschaften vorauszusagen. Der Computer setze die Atome zusammen wie Legosteine, erklärt der Berner Forscher. Er erzählt gern, wie er auf diesem Weg ein erstes «schönes Molekül» aus drei ineinander verschachtelten, ringförmigen Norbornanen entdeckte – einer Stoffklasse bestehend aus nur zwei Sorten von Atomen. Zu dieser gehört etwa das in Erkältungssalben verwendete Kampfer.
Der Aufbau der Datenbanken dauerte Jahre. Die verfügbaren Rechnerkapazitäten beschränkten die maximale Molekülgrösse auf zunächst 11 Atome. In der zweiten Datenbank lag die Grenze bei 13 und zuletzt bei 17 Atomen. Zugelassen waren dabei nur Elemente wie Kohlenstoff, Wasserstoff, Sauerstoff, Stickstoff und Fluor. Die Moleküle mussten ausserdem einfache Grundregeln der chemischen Stabilität einhalten: Vorgegeben sind Arten der Bindungen und gewisse Winkel zwischen den Bindungen.
KI sucht Kristalle
Trotz dieser Beschränkungen schwoll die Zahl der Moleküle auf unvorstellbare Grösse an. Das Legospiel des Computers führte schnell in die unendlichen Weiten des chemischen Raums. Reymond sah bald, dass es deutlich mehr unbekannte als bekannte Moleküle gab.
Die Generated Data Base GDB-11 zählte 26,4 Millionen Moleküle, GDB-17 ist mit 166 Milliarden Einträgen erwartungsgemäss die grösste Datenbank für kleine Moleküle weltweit. Allein um die Namen der enthaltenen Komponenten aufzulisten, braucht ein Rechner mehr als zehn Stunden. «Man darf sich nicht von der Menge der Moleküle blenden lassen», sagt Reymond. «Unsere Aufgabe ist es nicht, immer grössere Datenbanken zu bauen, sondern sie so zu gestalten, dass man darin suchen kann. Wir müssen die wenigen neuen Substanzen darin finden, die zu tatsächlich nutzbaren Strukturen führen. Es ist, als schürften wir nach Gold, und davon gibt es nicht endlos viel.»
Anatole von Lilienfeld von der Universität Basel durchforstet den chemischen Raum ebenfalls nach neuen, spannenden Verbindungen. Er sucht Elemente, die aus vier verschiedenen Elpasolith-Kristallen bestehen. Diese sind aufgrund ihrer Struktur komplexer als viele der üblichen Kristalle, die jeweils nur aus zwei oder drei Komponenten bestehen wie etwa das Kochsalz. «Elpasolithe haben interessante Materialeigenschaften, die sie zu Szintillatorkandidaten machen», sagt von Lilienfeld. Szintillatoren lassen sich anregen und leuchten dann in einem bestimmten Farbton.
Mit von künstlicher Intelligenz gestützten Verfahren entdeckte sein Team bislang 90 unbekannte Kristalle. Die Forschenden nutzten einen quantenmechanisch errechneten Datensatz von 10 000 Kristallen, um mit dem Modell die Eigenschaften von zwei Millionen weiteren Kristallen vorherzusagen. «Dank der künstlichen Intelligenz sparen wir zwei Millionen Franken an Rechenzeit», sagt Lilienfeld. Die Herstellung der Elpasolithe übernehmen Partner wie IBM Zürich oder das Swiss Nanoscience Institute in Basel.
Dank höherer Rechenleistungen und besserer Algorithmen können Chemikerinnen und Chemiker inzwischen mit Big-Data-Suchwerkzeugen und selbstlernenden Systemen die Schätze unter den Milliarden Verbindungen finden. Dadurch soll auch die Medikamentenentwicklung beschleunigt werden. Forschende können damit nämlich mögliche biochemische Prozesse komplett simulieren. Vor allem kleine Moleküle stehen im Fokus. Algorithmen erkennen, ob sie an bestimmte Proteine andocken und so aktiv werden können. Damit können sie die Datenbanken nach passenden Wirkstoffen durchforsten.
Ein Chemikerteam um Brian Shoichet von der University of California in San Francisco fahndete auf diese Weise in mehr als drei Millionen Substanzen nach einem neuen Schmerzmittel. Das Molekül sollte einen speziellen Opioid-Rezeptor aktivieren und somit Schmerz lindern, ohne dabei die üblichen Nebenwirkungen wie verringerte Atemfrequenz oder Verstopfung auszulösen. Der Algorithmus fand 23 Kandidaten, wovon sieben in ersten Labortests die gewünschte Wirkung zeigten. Derzeit versucht das von Shoichet gegründete Pharma-Start-up Epiodyne, daraus ein sicheres Medikament zu entwickeln.
Den chemischen Raum betreten
Zentral für solche erfolgreichen Wirkstofffahndungen sind verbesserte Werkzeuge, die es den Forschenden erlauben, sich in der wachsenden Datenwelt zurechtzufinden: Pubchem, eine Datenbank der amerikanischen National Institutes of Health, sammelt beispielsweise alle verfügbaren Informationen über mehr als 96 Millionen Moleküle. Surechembl verzeichnet rund 17 Millionen patentierte Verbindungen. In den vergangenen Jahren sind neben den drei GDBs von Jean-Louis Reymond zahlreiche solcher spezialisierten Verzeichnisse entstanden.
Reymond hatte sich eine Art Koordinatensystem des chemischen Raums ausgedacht. «Wir überlegten, welche einfachen Eigenschaften für das Verhalten eines Moleküls wichtig sind, und kamen nach einigem Herumprobieren mit dem System auf 42», erzählt der Chemiker. Jedes Molekül habe zählbare Eigenschaften wie die Zahl beteiligter Bindungen, der Ringstrukturen oder die Zahl und Art seiner Atome. Zusammen legen sie die Identität jedes Moleküls fest. «Ich bin selbst überrascht, dass das einfache System der 42 Dimensionen bis heute so schöne Ergebnisse liefert», sagt Reymond.
Jüngst publizierte Reymond mit seinem Doktoranden Daniel Probst eine neue Methode, um den 42-imensionalen chemischen Raum kleiner Moleküle in 2D- und 3D-Karten darzustellen, in denen man dann mit Virtual-Reality-Headsets herumwandern und die Strukturen erforschen kann. Diese Karten verdichten die wesentlichen Informationen über die Moleküle und stellen Unterschiede in ihren Wirkstoffkomponenten visuell dar.
Die Chemiker organisierten zunächst die Moleküle der Wirkstoffdatenbank Drugbank nach Grösse und Struktureigenschaften wie Steifigkeit oder elektrischer Polarität. «So entsteht eine Art Schattenspiel, bei dem ähnliche Moleküle in Clustern zusammenfasst sind», sagt Probst. Von bewährten Wirkstoffen aus kann man so nach ähnlichen Nachbarn suchen und per Mausklick visualisieren lassen. «Es geht darum, neue Ideen für Moleküle zu entwickeln», sagt Reymond. Gelingt das, haben die Chemiker im unendlichen Universum der Moleküle tatsächlich eine Goldader gefunden.
Hubert Filser arbeitet regelmässig für die Fernsehsendung Quarks & Co und wohnt in München.



